Alpha diversity
Wisam
2022-02-12
Part 1: Alpha diversity main measures.
dataset: phyloseq
The diet swap data set represents a study with African and African American groups undergoing a two-week diet swap. For details, see https://www.nature.com/articles/ncomms7342.
phyloseq-class experiment-level object
otu_table() OTU Table: [ 130 taxa and 222 samples ]
sample_data() Sample Data: [ 222 samples by 8 sample variables ]
tax_table() Taxonomy Table: [ 130 taxa by 3 taxonomic ranks ]Boxplots
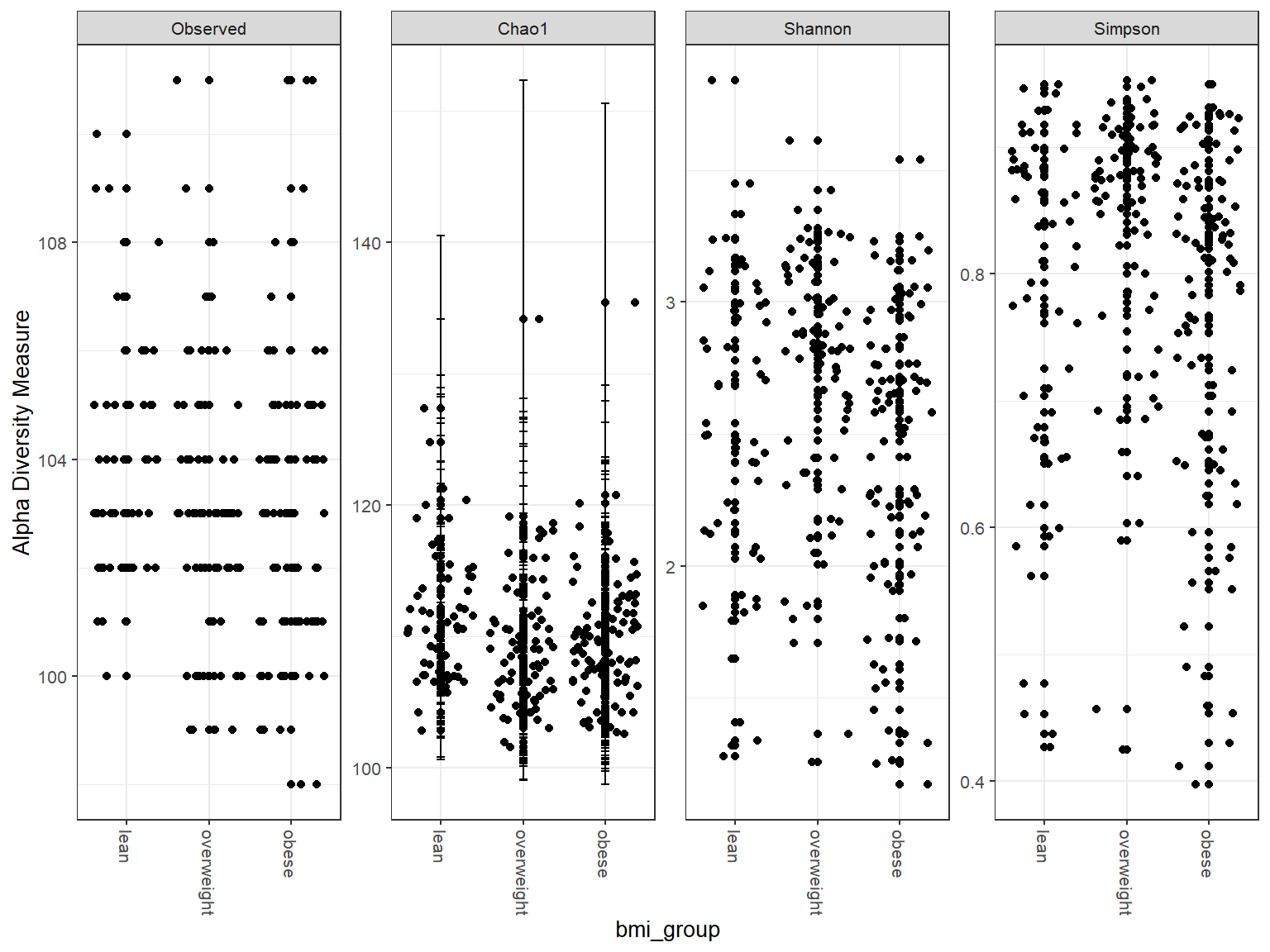
Violin plots
theme_set(theme_bw(10))
plot_richness(phy, x = "bmi_group", measures = alpha_metrics) + geom_violin(aes(fill = factor(bmi_group)),
trim = FALSE, draw_quantiles = c(0.25, 0.5, 0.75)) + geom_jitter(height = 0,
width = 0.1)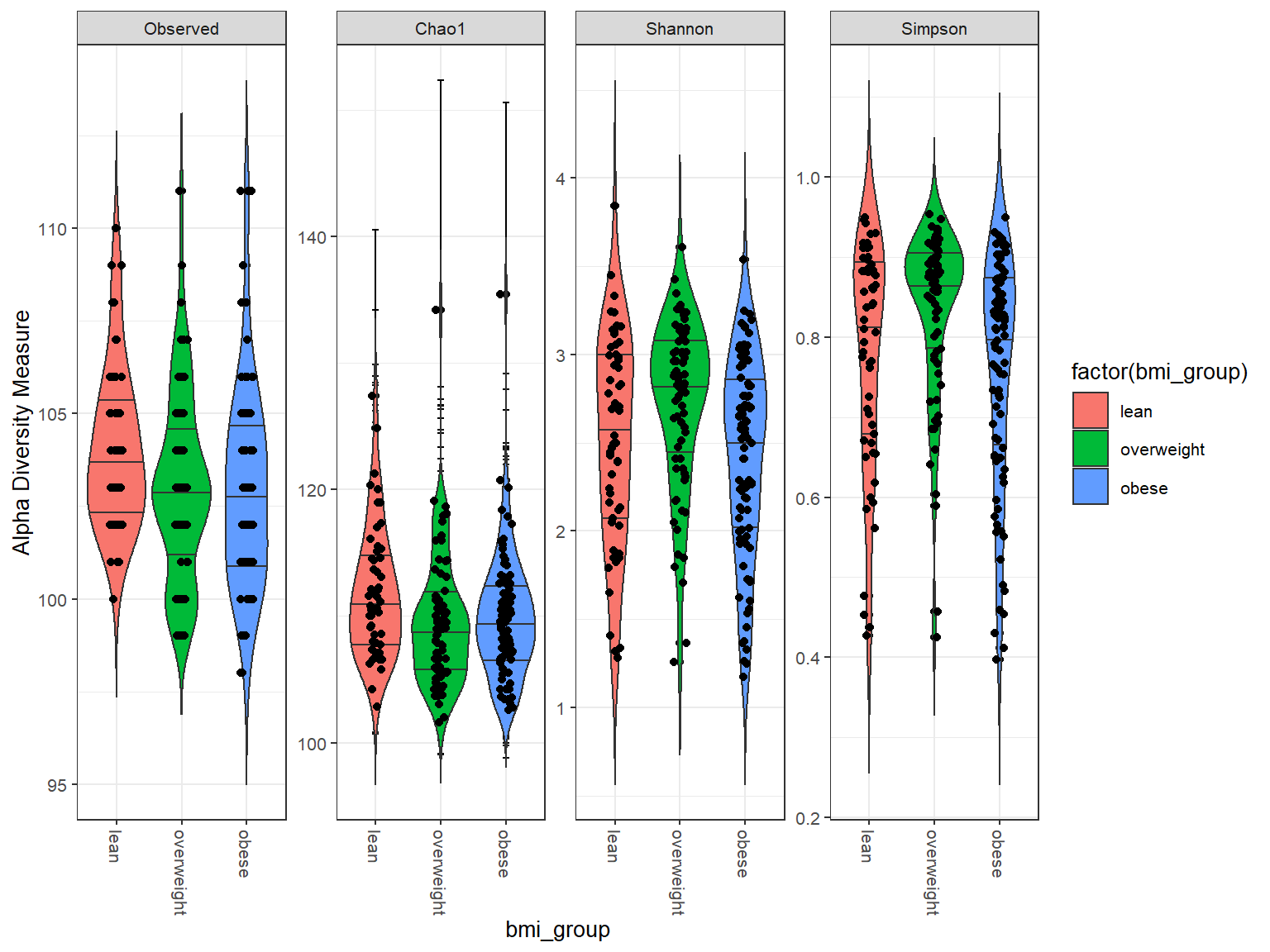
Part 2: non-parametric test
Kruskal-Wallis test
perform Kruskal-Wallis test between bmi_groups, since it has three factors. The purpose behind the test would be more clear.
alphas <- estimate_richness(phy, measures = alpha_metrics)
kw <- t(sapply(alphas, function(x) unlist(kruskal.test(x ~ sample_data(phy)$bmi_group)[c("p.value",
"statistic")])))
kw p.value statistic.Kruskal-Wallis chi-squared
Observed 0.022081536 7.626027
Chao1 0.015152446 8.379187
se.chao1 0.025379869 7.347598
Shannon 0.001053162 13.711917
Simpson 0.001623541 12.846291As Kruskal-Wallis test is significant
Post hoc
Post hoc by implementing Conover’s test
Using shannon index
df <- cbind(get_variable(phy), alphas$Shannon)
library("conover.test")
con <- conover.test::conover.test(df$`alphas$Shannon`, df$bmi_group, kw = T, method = "bonferroni") Kruskal-Wallis rank sum test
data: x and group
Kruskal-Wallis chi-squared = 13.7119, df = 2, p-value = 0
Comparison of x by group
(Bonferroni)
Col Mean-|
Row Mean | lean obese
---------+----------------------
obese | 1.177492
| 0.3604
|
overweig | -2.205510 -3.779721
| 0.0427 0.0003*
alpha = 0.05
Reject Ho if p <= alpha/2Using Simpson index
df1 <- cbind(get_variable(phy), alphas$Simpson)
con1 <- conover.test::conover.test(df1$`alphas$Simpson`, df1$bmi_group, kw = T, method = "bonferroni") Kruskal-Wallis rank sum test
data: x and group
Kruskal-Wallis chi-squared = 12.8463, df = 2, p-value = 0
Comparison of x by group
(Bonferroni)
Col Mean-|
Row Mean | lean obese
---------+----------------------
obese | 1.099905
| 0.4089
|
overweig | -2.162227 -3.646024
| 0.0475 0.0005*
alpha = 0.05
Reject Ho if p <= alpha/2Part 3: Linear regression
Using another phyloseq dataset from micorobiome package, this phyloseq has continuous variable among its metadata variables. We shall see the linear regression for the diversity_shannon as a dependent variable on the ‘age’ as the independent variable.
atlas1006
This data set contains genus-level microbiota profiling with HITChip for 1006 western adults with no reported health complications, reported in Lahti et al. (2014) https://doi.org/10.1038/ncomms5344.
phyloseq-class experiment-level object
otu_table() OTU Table: [ 130 taxa and 1151 samples ]
sample_data() Sample Data: [ 1151 samples by 10 sample variables ]
tax_table() Taxonomy Table: [ 130 taxa by 3 taxonomic ranks ]Linear regression model.
Analysis of Variance Table
Response: diversity$Shannon
Df Sum Sq Mean Sq F value Pr(>F)
age 1 3.167 3.1666 18.406 1.943e-05 ***
Residuals 1093 188.045 0.1720
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1It is significant p_value
Regression plot
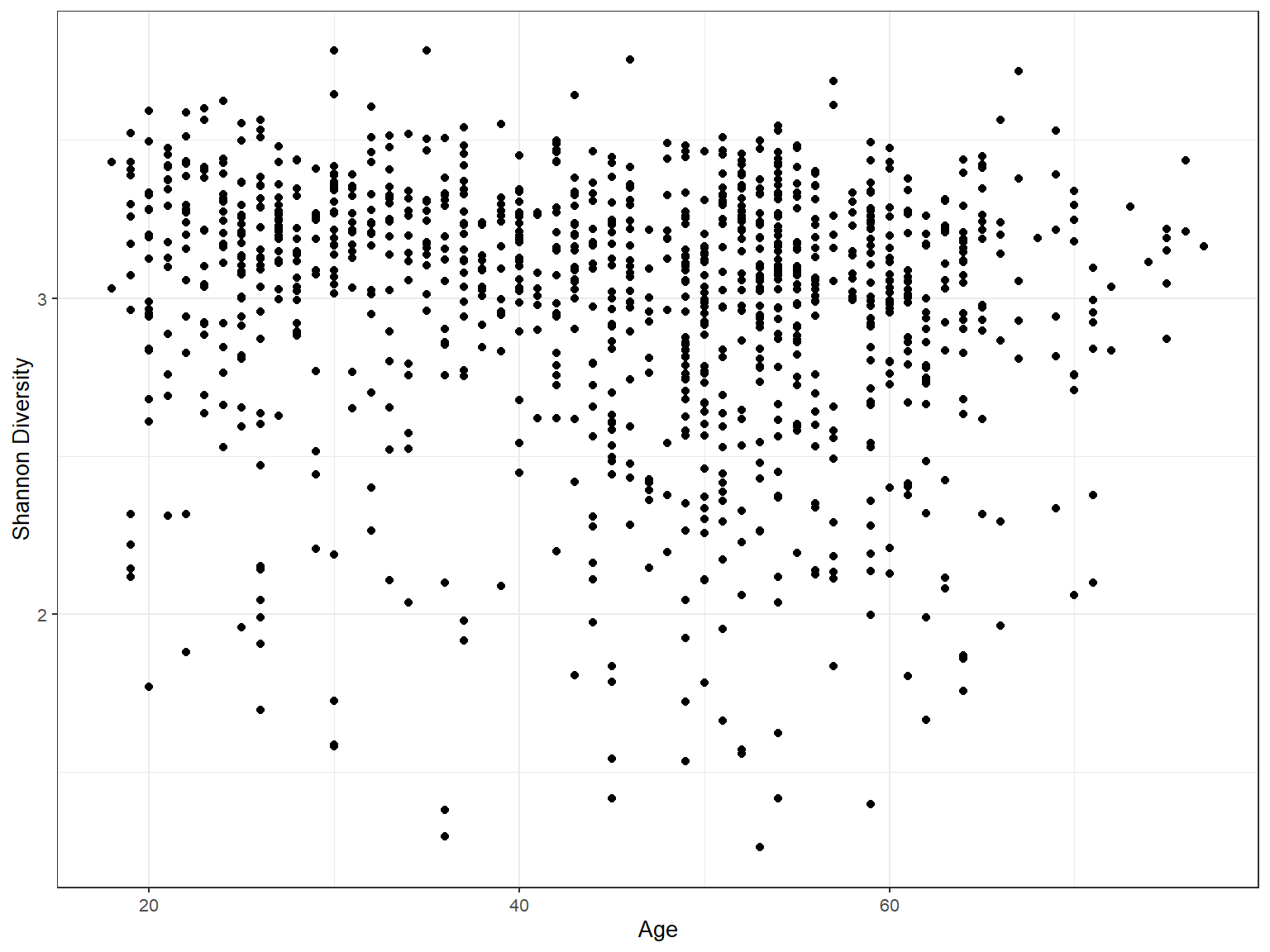
Acknowledgement An Introduction to Statistical Learning
regplot = function(x, y, ...) {
fit = lm(y ~ x)
plot(x, y, ...)
abline(fit, col = "red")
}
attach(data)
regplot(x = age, y = diversity$Shannon, xlab = "Age", ylab = "Shannon Diversity",
pch = 20)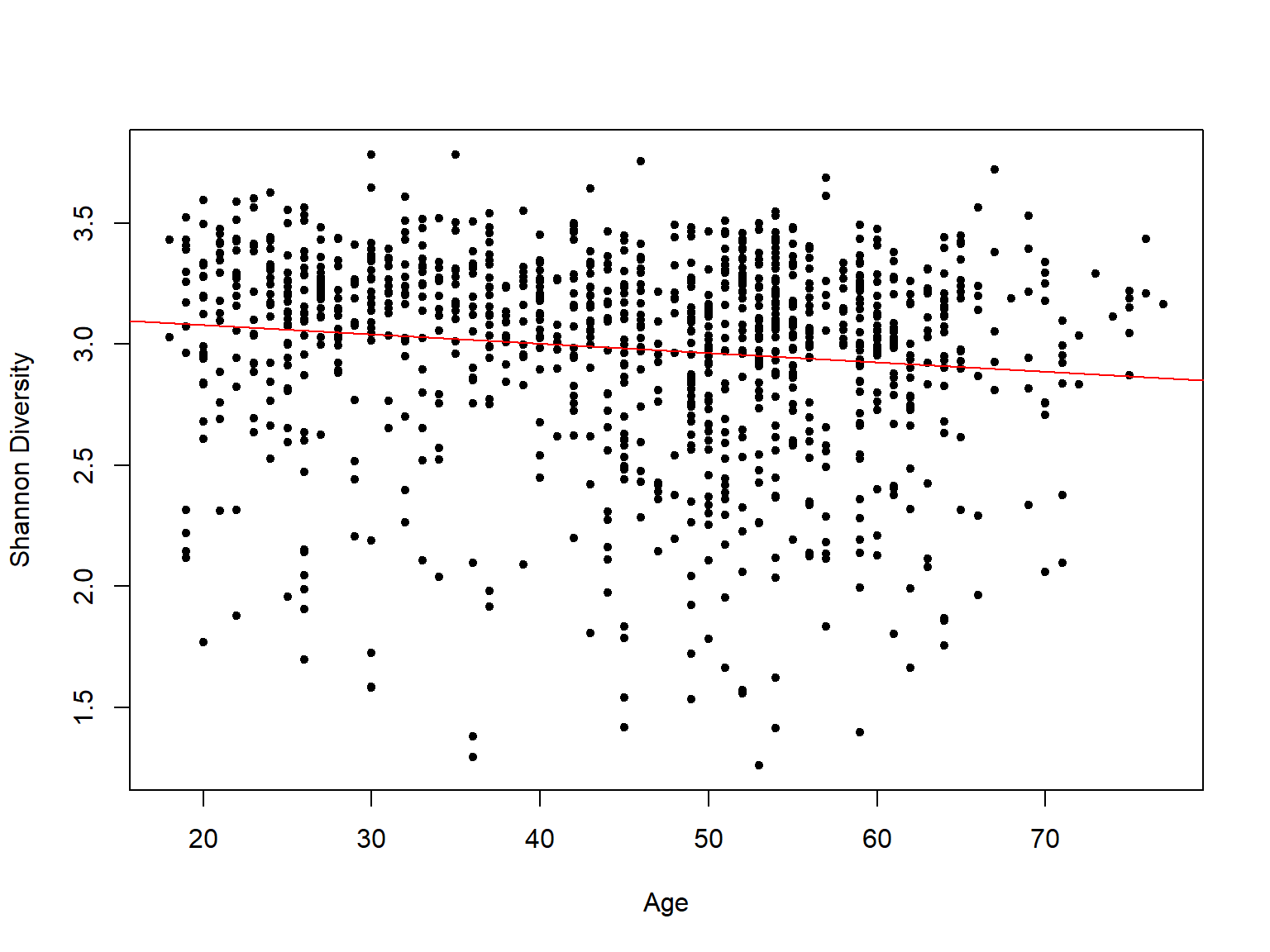 Important In microbiome analysis we do not check the normality conditions by Shapiro test for example, it is enough to check the histograms and see the pattern of the continuous variable.
Important In microbiome analysis we do not check the normality conditions by Shapiro test for example, it is enough to check the histograms and see the pattern of the continuous variable.